fastq文件的介绍
详细介绍:fastq格式
fastq是一种除了fasta文件之外,做生信另外一个最为常见的文件类型。
fastq文件主要是用来存储测序的序列信息的。它以4行为一个单位,是严格按照行限定的格式,不像之前的fasta文件那样比较灵活的形式。
示例
1 2 3 4 5 6 7 8 @SRR2177462.1 FCC600JACXX:2:1101:1489:2045/1 NGGCAAAAGGAAGCACATATTCGCATATAGAACCAGGATTTATAAGGTACAACAANTAGACTTATCCTCCACTCTCATGTTCATGAATCA + #1=ABDD?FH?HFG>DGHBHIFFHGCGGCHGCGHGII)?FG@DBAEH9???FGAB#-5@(@=EEHECAAH@EE;BCEEFA@ADDCCA;AC @SRR2177462.2 FCC600JACXX:2:1101:1661:2085/1 NAATGAAATTAAAGATAGCTGATCTATATTTCTCAAGTGACTAAGTATTAATATTATGCGTACTCTGTATTTCTCTAGTTGGTGGTTTAG + #4=DDFFFHHFHFIGIHIIIJEGHIGIAHIJFIEHHHIIIIIEIHIIIJCFIHJIHIIAHIFFGIIJHFHIIJIJF@FEHGIDHHA?B@C
现在我们从头开始,每4行为一组,在每一组之内的:
1 @SRR2177462.1 FCC600JACXX:2:1101:1489:2045/1
第一行 :以@字符开头,表示这条序列的测序信息,来自于哪台测序仪、第几个run、第几个lane、第几个tail、以及在这个tail的X轴与Y轴的位置。
1 NGGCAAAAGGAAGCACATATTCGCATATAGAACCAGGATTTATAAGGTACAACAANTAGACTTATCCTCCACTCTCATGTTCATGAATCA
第三行 :以+字符开头,后面跟着ID序列标识符,如果+后面有东西那么必须与第一行一致,一般都是没有的,为了节省内存。
1 #1=ABDD?FH?HFG>DGHBHIFFHGCGGCHGCGHGII)?FG@DBAEH9???FGAB#-5@(@=EEHECAAH@EE;BCEEFA@ADDCCA;AC
第四行 :是第二行序列每个碱基的质量编码值,包含与第二行字符数量相同的符号。经过转换之后可以得到这个碱基的质量数值,以及这个碱基的错误率。
前期准备
因为一般的fastq文件都是经过压缩的,如果你想拿fastq的实际文件来做这次的测试,那样可能比较耗时间,特别是在gzip或者pigz的解压时间上面。所以这里如果你想快速进行实战演练的话,我建议先把本文末尾的测试数据这一步做了,得到123.fastq.gz进行测试会快一些。
注意!!
由于我演示是使用的windows系统,在文件的一行结束的位置除了一个\n换行符之外,还有\r回车符这样的字符存在,而使用perl中的chomp方法不能除去\r回车符,所以下面代码中,在Mac或者Linux中可以直接写为chomp我换成了s/\r?\n//。在后面生成123.fastq.gz文件的时候粘贴复制到文本里面去的时候也可能带上回车符\r,这里使用windows进行测试要注意了。
获取fastq文件的信息
1. 序列条数
思路 : 利用fastq的四个为一个单位的特征
1 2 3 4 5 6 7 zcat 123.fastq.gz | perl -n -e ' $n++; # 因为四个为一组,没必要将所有的行都加起来再除以4 # 消耗掉其他三行 <>;<>;<>; END{print $n} '
1 2 3 4 5 6 zcat 123.fastq.gz | perl -n -e ' if($. % 4 == 1){ $n++; } END{print $n} '
或者使用pigz,pigz要比gzip解压快一些,但是git for windows上没有这个东西,另外使用zcat进行查看文件我发现比直接解压文件要快,所以后面测试还是使用zcat。不过也展示一下pigz的使用方式:
1 2 3 4 5 6 7 pigz -d -c 123.fastq.gz | perl -n -e ' $n++; <>;<>;<>; END{print $n} '
1 2 3 4 line=`zcat 123.fastq.gz | wc -l` let seq_num=$line /4echo $seq_num
输出为
2. 质量值的类型
有关质量值的编码方式
现在一般都是采用的Phred+33类型的,那为什么还要说这一小节呢?
通过介绍这些质量值类型可以帮助理解fastq中质量值与序列的关联性以及在这种利用文本传达信息的过程中,人们所展现出来的智慧!
测序的错误来源
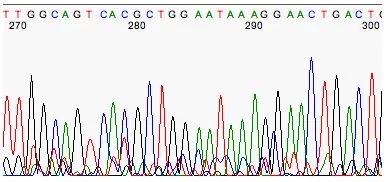
1 2 3 4 5 6 7 8 +==============+ +==============+ | . . . .| | * * * *| | . . . | ==> | * * * | | . . . .| | * * * *| | . . . | | * * * | +==============+ +==============+ 测序芯片中测序点 激发之后产生荧光
在测序的时候,是通过对应位置发出来的对应颜色的荧光的强度来得到这个位置测定出。
那测序的错误是从何而来呢?
在荧光染料测序中,每次发生碱基合成时会释放出荧光信号,该信号被CCD图像传感器捕获。记录下荧光信号的峰值,生成一个实时的轨迹数据(chromatogram)(如上图所示)。因为不同的碱基用不用的颜色标记,检测这些峰值即可判断出对应的碱基。但由于这些信号的波峰、密度、形状和位置等是不连续或模糊的,有时很难根据波峰判断出正确的碱基。通过对每一个点对不同颜色的曲线参与的程度进行计算,计算许多与波峰大小和分辨率相关的参数,根据这些参数,从一个巨大的查询表中找出碱基质量得分。这个查询表是根据对已知序列的测序数据分析得到的(应该是分析得到波峰参数与碱基错误率的关系,再通过公式把错误率转换成质量得分,得到波峰参数与质量得分的直接对应表)。
波长1
波长2
波长3
波长4
A
0.65
0.14
0.01
0.02
B
0.81
1.03
0.02
0.03
C
0.02
0.02
0.95
0.04
D
0.03
0.03
1.10
1.56
表2.2.0.1 例如一个测序位点不同碱基所带的荧光基团在这个点发出的荧光的贡献
那么也就是说,一个点ATGC四个碱基上经过激发之后得到不同的波长,如果这个位点不同颜色占据的量越多,那么出错的可能性就越大,如果某种颜色占据的越多那么越有可能是这个碱基。那么如何描述这种碱基的纯粹程度呢?
错误的评判标准
Quality Score —— 评判一个碱基判读可靠性指标
Q = − 10 l o g ( P ) Q = -10log(P)
Q = − 1 0 l o g ( P )
准确度
P值(错误率)
Q值(质量值)
90%
10%
10
99%
1%
20
99.9%
0.1%
30
99.99%
0.01%
40
99.999%
0.001%
50
平常常常说Q30,就是说碱基准确度达到99.9%的程度,就是说出错的可能性为0.1%。这里别人推荐了一种记忆方法,10是一个9,20两个9,40是4个9。
这里有关的测序步骤以及质量值可以参看陈老师的视频
陈巍学基因
有关P值更加详细的计算说明见这篇文章P值与基因组学(1):从fastq文件的分析的分析谈起
那么就需要对每次每个测得的碱基的质量进行记录,怎么来记录呢?可是质量值从个位数到十位数会发生什么呢?
质量值的记录方式
使用数值的序列与质量值得对应关系
本来对应关系是这样的
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 N 2 A 19 A 28 T 35 G 35 A 37 A 37 A 37 T 39 T 39 A 37 A 39 A 37 G 40 A 38 . . . . . .
写到一行上(其实没有这种数值类型的表示方式)
1 2 3 序列 NAATGAAATTAAAGATAGCTGATCTATATTTCTCAAGTGACTAAGTATTAATATTATGCGTACTCTGTATTTCTCTAGTTGGTGGTTTAG 质量 21928353537373739393739374038403940404041363839403840323940413740363939394040404040364039404040413437403941403940403239403737384040413937394040414041373137363938403539393230333134
就单单只看两个碱基,碱基与质量值之间的对应关系有多种
1 2 3 4 5 6 7 8 9 10 11 N => 2 A => 1 或者 N => 21 A => 9 或者 N => 2 A => 19 或者 N => 21 A => 92
如果这么写的话,第一个碱基质量值为个位数,其他的为十位数,这样记录之后,后续程序怎么来分析它呢?一个碱基对应的质量值到底是个位数还是十位数呢?这个样子质量值与碱基之间怎么对应啊?这是个问题…
那如果用空格将数值间隔开呢?
1 2 序列 NAATGAAATTAAAGATAGCTGATCTATATTTCTCAAGTGACTAAGTATTAATATTATGCGTACTCTGTATTTCTCTAGTTGGTGGTTTAG 质量 2 19 28 35 35 37 37 37 39 39 37 39 37 40 38 40 39 40 40 40 41 36 38 39 40 38 40 32 39 40 41 37 40 36 39 39 39 40 40 40 40 40 36 40 39 40 40 40 41 34 37 40 39 41 40 39 40 40 32 39 40 37 37 38 40 40 41 39 37 39 40 40 41 40 41 37 31 37 36 39 38 40 35 39 39 32 30 33 31 34
有没有感觉到瞬间多了好多字符一样的,这样带来的后果就是占用的内存急剧增加。
于是乎人们就想到了一个好办法
使用ASCII码的序列与质量值得对应关系
1 2 序列 NAATGAAATTAAAGATAGCTGATCTATATTTCTCAAGTGACTAAGTATTAATATTATGCGTACTCTGTATTTCTCTAGTTGGTGGTTTAG 质量 #4=DDFFFHHFHFIGIHIIIJEGHIGIAHIJFIEHHHIIIIIEIHIIIJCFIHJIHIIAHIFFGIIJHFHIIJIJF@FEHGIDHHA?B@C
可以看到一个碱基对应一个表示质量值的字符,这样既减少了文件所占据的内存,也让碱基质量值与碱基的对应关系更加清晰。
那么将使用ASCII码进行记录的质量值与直接使用数字进行记录的质量进行比较,使用数字表示法:
文件占据内存增加 :在fastq文件中,占内存的大头是序列以及质量值的文字,如果质量值用两个数字表示那内存占据会增加很多碱基与质量值对应的关系 :如果是以数字的形式对应碱基的话,如果一直是1个碱基对应2个数字那都好说,可是保不齐某些碱基测序质量极差,质量值为个位数,那么既有1对2、也有1对1,怎么才能确定碱基与质量值之间的对应关系呢?
既然采用ASCII码来记录碱基质量,那么为什么还要分什么Phred+33 、Phred+64 之类的?
质量值的编码方式
现有的常见的测序质量值编码方式
1 2 3 Quality encoding: !"#$%&'()*+,-./0123456789:;<=>?@ABCDEFGHIJ | | | | || Quality score: 0........10........20........30........4041
1 2 3 Quality encoding: @ABCDEFGHIJKLMNOPQRSTUVWXYZ[\]^_`abcdefgh | | | | | Quality score: 0........10........20........30........40
1 2 3 Quality encoding: ;<=>?@ABCDEFGHIJKLMNOPQRSTUVWXYZ[\]^_`abcdefgh | | | | | | Quality score:-5....0........10........20........30........40
话说Phred 和solexa 是什么意思呢?百度Phred 之后发现有一个很古老的网站 ,网页比较简洁,里面写着
Phred is a base-calling program for DNA sequence traces. Phred reads DNA sequence chromatogram files and analyzes the peaks to call bases, assigning quality scores (“Phred scores”) to each base call.
就是说Phred 这个原来是一个工具,用来对测序得到的chromatogram文件分析(可以见上面的有四条曲线的图)。这个软件通过对四种不同的荧光的强度来分析得到该碱基位点的质量值。
当时是1998年发现核酸研究上的一篇文章《 Base-calling of automated sequencer traces using phred. I. Accuracy assessment》然后同年又发了文章《Base-calling of automated sequencer traces using phred. II. Error probabilities》。
最初开发时,Phred在所检查的数据集中产生的错误明显少于其他方法,平均减少40-50%的错误。Phred质量评分已被广泛接受用于表征DNA序列的质量,并且可用于比较不同测序方法的功效 。Phred在人类基因组计划中发挥了重要作用,自动脚本处理大量序列数据。它当时是学术和商业DNA测序实验室最广泛使用的碱基调用软件程序,因为它具有很高的碱基调用准确性。 ——wikipedia
而solexa 是一种基于边合成边测序的技术。通过单分子阵列实现小型阵列上的桥式PCR反应。新的阻断技术能够每次只合成一个碱基,经过激光激发之后得到激发光,对激发光进行分析得到碱基信息。
早些年,人们在谈到新一代测序仪时,经常会提起Solexa,而不是Illumina。这是一家低调的公司,规模也不大,但是测序技术却非常新颖。它开发出的测序仪,在通量上领先于其他竞争产品。收购Solexa,也成为Illumina的转折点,从此踏上高速发展的道路。 ——生物通
现在大部分都是用的Illumina 1.8+的Phred+33方式,也就是将质量值加上33,得到一个数值,再将这个数值按照上面的ASCII码表对应到特定的字符上去。
比如
Q值(质量值)
+33之后的值
对应的字符
0
33
!
1
34
"
10
43
+
20
53
5
30
63
?
40
73
I
41
74
J
为什么偏要+33,而不是+11啊!+22啊之类的?
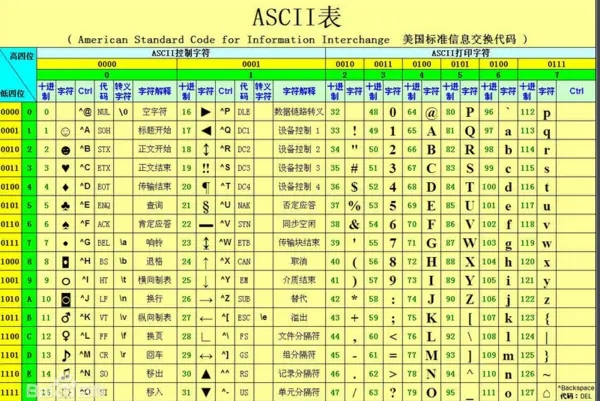
你可以看看上面的ASCII码表,因为在0~32这个范围之内有很多奇形怪状的字符,同时还包含什么制表符、换行符、回车符、空格、响铃、退格等这类的看不见的字符。而很多时候质量值需要一是肉眼能方便查看;二是要方便程序读取。显然在0-32这个范围内不适合取。而从33开始直到127均为可见的字符。那从33开始取值不就的了,怎么又来个什么+64之类的?
谷歌了一下,暂时没有找到究竟+64的原因,但是在一篇博客《质量值体系 Phred33 和 Phred 64 的由来 及其在质量控制中的实际影响 - Part 1》中作者自己脑补的两者之间的关联。我觉得下面这句话可能可以说明一下作者脑补的内容:
一流的企业做标准,二流的企业做品牌,三流的企业做产品
就是当时测序公司有多家,互相之间相互竞争,各家公司是竞争对手,怎么可能你用什么我也用什么。所以我也要想出一定的标准,哈哈!后来的Solexa就不采用+33的方式(这个方式就是Phred+33的编码方式当时Sanger公司所采用的),而使用Solexa+64 ,其实+64也不是就是随便来的,可以再看一下上面的ASCII码对照表。
1 2 3 Quality encoding: @ABCDEFGHIJKLMNOPQRSTUVWXYZ[\]^_`abcdefgh | | | | | Quality score: 0........10........20........30........40
在质量值0对应的@之后,就是从英文大写字母A开始了,也就是质量值1-26对应编码字符A-Z了。这样人肉眼看的话其实质量值是相当直观了。不过在质量值到达33之后,又出现了小写字母…这个个人感觉就不如Phred+33那样的呢。
到后来2006年Solexa公司被Illumina公式收购,于是又有了Illumina 1.3+ Phred+64(这里是我自己脑补的- . -)。于是就有了Sanger公司的Phred+33的方式以及Illumina的Phred+64的方式。于是两家公司互相不让着谁。这可苦了当时编写生信数据分析的人,后来没办法只有在程序中加上自动判断的程序。
可是墨菲他老人家不高兴了(这里我不太了解墨菲和这个事件之间的联系),所以在 2011 年, Illumina 公司表示他们又要改成 Phred33 体系了 (Upcoming changes in CASAVA )。这样来来回回还是回到了Phred+33的体系。
期间有意思的是,当时三巨头的另一家测序仪公司 454 Life Sciences (后被 Roche 收购) 就更绝, 人家从碱基开始就不用 ACTG 表示, 直接整了个 ColorSpace 体系出来, 根本不和你们玩,当然后来大家也不跟 454 玩了, 最后他也就没得玩了 。
这个是在网上别人得到的 ColorSpace数据:
1 2 3 4 5 6 7 8 @SRR2967009.1 100_1000_1168_F3 T10011023211201220121202030102221012302121010131001 + 2@@@@>@?@@@@<@@//;@@/@9?@8@=@@@6;6@66;<@6@67?2?;/@ @SRR2967009.2 100_1000_1211_F3 T20132312201120021312220200023110220113100012321011 + @@@@@@@@@<@@@@@@@@@@@@@@@@@@@@@@?@@@@/?@@@@@@@@<?@
序列是用double-encoded data或者称colorspace的模式表示的。
按照手册的说明:
1 2 3 4 AA, CC, GG, TT : 0 AC, CA, GT, TG : 1 AG, CT, GA, TC : 2 AT, CG, GC, TA : 3
Therefore five base long sequence AACTA will be represented as 0123. The encoding is AA=0, AC=1, CT=2, and TA=3.
额,感觉有些诡异。不过这样做应该有他的考虑,只是现在我不清楚。
质量值的区别方法
肉眼查看 [0~9]的为Phred33特有的质量字符,小写字母[a~z]的为Phred64特有的质量字符。程序判断
这个shell代码不知道出处是哪里,反正放在这里吧,如果有侵权,那我就删除了。
1 2 3 4 5 6 7 8 9 10 11 fqtype less $1 | head -n 999 | awk '{if(NR%4==0) printf("%s",$0);}' \ | od -A n -t u1 -v \ | awk 'BEGIN{min=100;max=0;}\ {for(i=1;i<=NF;i++) {if($i>max) max=$i; if($i<min) min=$i;}}END\ {if(max<=74 && min<59) print "Phred+33"; \ else if(max>73 && min>=64) print "Phred+64"; \ else if(min>=59 && min<64 && max>73) print "Solexa+64"; \ else print "Unknown score encoding"; \ print "( " min ", " max, ")";}' }
仿照上面的shell代码,我改成了Perl One-liners的版本。一般只需要取一部分序列进行计算就可以了,计算这个值还是挺快的。使用真实的fastq数据同样也很快,只需要接近0.05秒的时间。(😒你可能会说你看上面的bash脚本就这么几行就完事了,怎么用perl要写这么多!你之前应该也听说过perl有一句名言事实的真相不止一个!,如果把perl写成很单行 的形式,估计不好看懂,这里是为了展示代码的转换时是个什么原理,而且代码如果是自己用,不要装逼到吓到自己 !)
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 test_line=80 zcat 123.fastq.gz | head -n $test_line | perl -n -e ' BEGIN{ # 首先初始化两个用来判断质量值上下限的两个变量 $max = 0; # 将最大值设置得精良小 $min = 1000; # 将最小值设置得尽量大 } # 质量行在4的倍数行 if($. % 4 == 0){ # 去除回车符换行符 s/\r?\n$//; my $word; # 每次“吞”一个字符进行ASCII码值转换 while($word = chop($_)){ my $ascll_num = ord($word); # 不断扩大min与max的范围,得到更加真实的上下限值 if($ascll_num > $max){$max = $ascll_num;} if($ascll_num < $min){$min = $ascll_num;} } } END{ # 如果质量最大值低于75,最小值小于59,那么认为就是Phred+33类型 if($max < 75 and $min < 59){ print "Phred+33!\n"; }elsif($max > 73 and $min > 63){ # 如果质量最大值高于73,最小值高于63,那么认为就是Phred+64类型 print "Phred+64!\n"; }elsif($min > 53 and $min < 64 and $max > 73){ # 如果最小值在58和64之间,最大值大于73,那就是Solexa+64类型 print "Solexa+64!\n"; }else{ # 否则不知道类型 print "unkown!\n"; } # 打印出上下限信息 print "Max:$max\n"; print "Min:$min\n"; } '
输出为:
但是这里你觉得这样的步骤可不可以分解呢?
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 test_line=80 zcat 123.fastq.gz | head -n $test_line | awk 'NR%4==2{print}' | perl -n -e ' BEGIN{ # 首先初始化两个用来判断质量值上下限的两个变量 $max = 0; # 将最大值设置得尽量小 $min = 1000; # 将最小值设置得尽量大 } # 去除回车符换行符 s/\r?\n$//; my $word; # 每次“吞”一个字符进行ASCII码值转换 while($word = chop($_)){ my $ascll_num = ord($word); # 将上下限的值赋值给刚才的两个变量 if($ascll_num > $max){$max = $ascll_num;} if($ascll_num < $min){$min = $ascll_num;} } END{ print "$max:$min\n"; } ' | perl -n -a -F: -e ' BEGIN{ sub print_type { my ($max,$min)=@_; if($max < 75 and $min < 59){ return "Phred+33!\n"; }elsif($max > 73 and $min > 63){ return "Phred+64!\n"; }elsif($min > 53 and $min < 64 and $max > 73){ return "Solexa+64!\n"; }else{ return "unkown!\n"; } } } my ($max,$min) = @F; print "Max:$max\n"; print "Min:$min\n"; '
3. fastqc相关统计量
序列长度的分布
这个统计序列长度就比统计fasta序列的长度简单一些啦!
1 2 3 4 5 6 7 8 9 10 11 12 13 14 zcat 123.fastq.gz | awk 'NR%4==2{print}' | perl -n -e ' s/\r?\n$//; my $seq_len = length($_); # 把序列长度以及为此长度的信息存入哈希中 $hash{$seq_len}++; END{ # 打印出不同长度以及为此长度的序列的条数 print "length\tcount\n"; for my $len (sort {$a <=> $b} keys %hash){ print "$len\t$hash{$len}\n"; } } '
输出为
1 2 3 4 length count 89 1 90 18 91 1
也可以重定向到文件中,拿这个文件可以使用R来画图。
同样的你也可以写成:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 zcat 123.fastq.gz | perl -n -e ' if($. % 4 == 2){ s/\r?\n$//; my $seq_len = length($_); # 把序列长度以及为此长度的信息存入哈希中 $hash{$seq_len}++; } END{ # 打印出不同长度以及为此长度的序列的条数 print "length\tcount\n"; # 按照长度进行排序 for my $len (sort {$a <=> $b} keys %hash){ print "$len\t$hash{$len}\n"; } } '
实际上上面的方法比下面的要慢一些,但是上面的程序将perl所承担的任务量减少了,所以代码量看起来少了一些。有的时候不能兼得,每项需要均衡一下。
放松时间
1 |_・) |ω・) |・ω・`) (╯ε╰) ^.^ (๑→ܫ←) ←_← (╯ε╰) ^.^ (๑→ܫ←) ←_← (╯ε╰) ^.^ (๑→ܫ←) ←_← ヘ(・_|
最近听了一下重阳世界观-狂点技能树(1),和大家分享一下,里面说到:
在兵马俑坑,出土最多的青铜兵器是箭头,考古人员发现了4万多支箭头,是三棱型的。制作的非常规范,箭头底部宽度的误差为0.83毫米,而且金属配比基本上是相同的。
为什么选择这种三棱形状的箭头呢?
三棱箭头拥有三个锋利的棱角,在击中目标的瞬间,棱的锋刃处就会形成切割力,箭头就能够穿透铠甲,直达人体。同时这三个面是呈流线型的,据说这个流线型的轮廓和子弹的外形都一致。按照现在话来说,有一个好的空气动力学的特性。除了三棱型的箭矢,还有什么狼牙箭——带倒钩和翼面的。但翼面容易受风的影响,使箭头偏离目标。
武器有很多个维度,如果只追求单一的维度,那么这个武器不会太好,就说这样一个小小的箭头啊!制作工艺简单,标准化,那样才能大量生产;如果太复杂,成本太高,不能大量装备;另外如果只盲目追求杀伤力,打不准那也不行。
三棱箭头.jpg
各种各样的箭头.jpg
每个位置上质量值的分布
其实在fastQC软件之中就对这个做了一个很好的诠释,这里我用Perl代码+R代码尝试实现这种每个位置上的碱基质量分布:
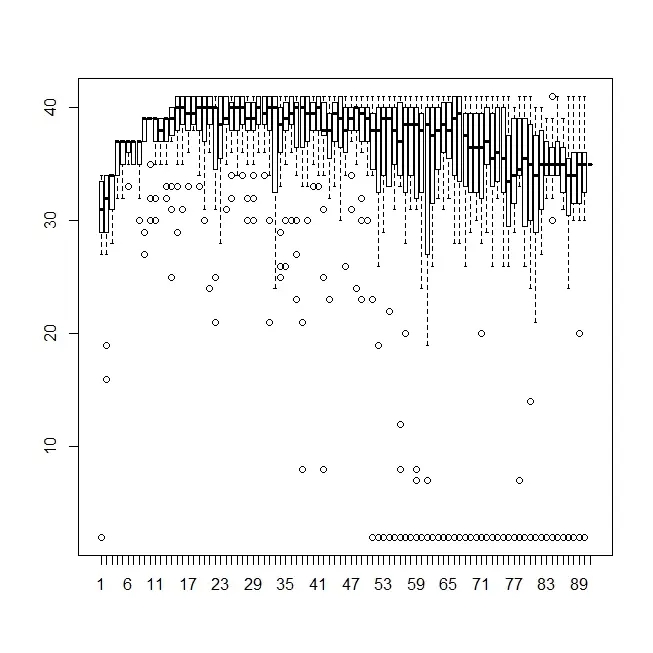
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 zcat 123.fastq.gz | perl -n -e ' BEGIN{ # 先声明一个质量值转换的子程序 sub convert_quality_to_score{ my $ascll = shift; # 这里我默认使用Phred+33 return ord($ascll) - 33; } } # 只需要每个单位的第四行 if($. % 4 == 0){ s/\r?\n//; my $n = 0; while($_){ $n++; my $quality_score = substr($_,0,1,""); # 将碱基推到对应的位置的哈西中 push @{$hash{$n}},convert_quality_to_score($quality_score); } } END{ my @list; # 设置列表内插之后打印元素的分隔符 $" = ","; for my $base_site (sort {$a <=> $b} keys %hash){ # 按照R语言的方式写入信息 print "c_${base_site} <- c(@{$hash{$base_site}})\n"; push @list,"c_${base_site}"; } # 写入执行箱线图的boxplot print "boxplot(@list)\n"; } ' > statistic_base_quality.RR source ("statistic_base_quality.R" )
就会得到如下的类似的箱线图,比较简陋啊,但是也算是有一种实现方式了,但是fastQC内部的实现机制是什么我现在还不知道。不过我感觉按照我这么写测序文件小可以用,大了估计不一定好使。
刚才在服务器上试了一下,发现这个运行的时间有点长…到时候运行估计会把R给卡死。算了!就当我这个例子没有举过v_v。
N值的统计
其实这个统计与之前的统计序列长度类似:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 zcat 123.fastq.gz | awk 'NR%4==2{print}' | perl -n -e ' s/\r?\n$//; my $word; my $n = 0; while($word = substr($_,0,1,"")){ $n++; $hash{$n}++ if uc($word) eq "N"; } END{ # 打印出不同长度以及为此长度的序列的条数 print "site\tcount\n"; for my $site (sort {$a <=> $b} keys %hash){ print "$site\t$hash{$site}\n"; } } '
这个文件也可以重定向生成tsv文件,之后可以用R语言读取画图
格式转换
1. fastq转换为fasta
网上一般使用的是perl的Bio::Perl模块来进行处理,其实这里不使用模块也可以进行转换。
普通转换
将第一行作为titie,第二行作为序列组成fasta文件,如果想要将序列分割,那么请看我之前的文章perl 命令行实战1 - fasta文件的相关操作,只需要用一个管道操作符将不同perl的小的单行程序连起来就可以了。
1 2 3 4 5 6 zcat 123.fastq.gz | awk 'NR%4==1{print};NR%4==2{print}' | perl -n -e ' (my $title = $_) =~ s/\r?\n//; $title =~ tr/@: />_-/; (my $sequence = <>) =~ s/\r?\n//; print "$title\n$sequence\n"; '
也可以把质量值保留下来
1 2 3 4 5 6 7 8 9 10 11 zcat 123.fastq.gz | awk 'NR%4!=3{print}' | perl -n -e ' (my $title = $_) =~ s/\r?\n//; $title =~ tr/@: />_-/; (my $sequence = <>) =~ s/\r?\n//; (my $quality = <>) =~ s/\r?\n//; print "$title\n$sequence\n"; # 将质量值信息追加到文件中 open FILE,">>","123.fasta.quality" or die $!; print FILE "$title\n$quality\n"; close FILE; '
而这个fasta序列你可以用重定向的方式输出到文件中去或者进入管道继续处理
特殊转换
有的时候有些软件需要fasta的文件名比较特殊,比如>infile_0/1/0_1234,这样的之类的,
1 2 3 4 5 6 7 8 9 zcat 123.fastq.gz | awk 'NR%4==2{print}' | perl -n -e ' $n++; # 变量内插一下就完事儿了 my $title = ">infile_0/1/0_$n"; (my $sequence = $_) =~ s/\r?\n//; print "$title\n$sequence\n"; '
质量编码标准的转换
0. 首先要确定质量值编码方式
如果不知道质量值的编码方式胡乱的转可能会出问题!或者要么写一个脚本先自动判断质量值类型,然后再转换。
可以先按照2.2 质量值的类型这一步进行测试,或者已知质量值,然后再进行转换。
现在手头上没有相应的文件,只能说在这里写一下代码吧
1. Phred+64转换为Phred+33
思路 :+64变成+33,怎么办呢?减去32?试一下
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 zcat 123.fastq.gz | perl -n -e ' BEGIN{ sub convert_64_to_33 { my $word = shift; return chr(ord($word) - 32); } } print $_; (my $sequence = <>) =~ s/\r?\n//; (my $title2 = <>) =~ s/\r?\n//; (my $quality = <>) =~ s/\r?\n//; my $word; my $quality_new; while($word = chop($quality)){ $quality_new = convert_64_to_33($word) . $quality_new; } print "$sequence\n"; print "$title2\n"; print "$quality_new\n"; '
你也可以把chr(ord($word) - 32);这一句话改为chr(ord($word) + 32);,你觉得应该变成什么功能呢?
2. Solexa+64转换为Phred+33
思路 :由于Solexa的质量值计算方式与Phred方式不同,所以需要对质量值计算标准做一个转换。这里参考了Perl中FastQ与FastA格式的相互转换
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 zcat 123.fastq.gz | perl -n -e ' BEGIN{ # 下面的算法参考自博文《Perl中FastQ与FastA格式的相互转换》,在这里还是说明一下 my @conv_table; for (-64..64) { $conv_table[$_+64] = chr(int(33 + 10*log(1+10**($_/10.0))/log(10)+0.499)); } sub convert_Solexa64_to_Phred33{ my $word = shift; return $conv_table[ord($word)]; } } print $_; (my $sequence = <>) =~ s/\r?\n//; (my $title2 = <>) =~ s/\r?\n//; (my $quality = <>) =~ s/\r?\n//; my $word; my $quality_new; while($word = chop($quality)){ $quality_new = convert_Solexa64_to_Phred33($word) . $quality_new; } print "$sequence\n"; print "$title2\n"; print "$quality_new\n"; '
筛选fastq文件的序列
1. 按照长度进行筛选
长度筛选类似于fasta文件的长度筛选,而且还简单一些
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 export upper_limit=89export lower_limit=89zcat 123.fastq.gz | perl -n -e ' # 将每一行末尾的回车符换行符去掉 (my $title = $_) =~ s/\r?\n//; (my $sequence = <>) =~ s/\r?\n//; (my $comment = <>) =~ s/\r?\n//; (my $quality = <>) =~ s/\r?\n//; # %ENV是用来存周围环境中的变量 if(length($sequence)>=$ENV{lower_limit} && length($sequence)<=$ENV{upper_limit}){ print "$title\n$sequence\n$comment\n$quality\n"; } '
输出为
1 2 3 4 @SRR2177462.1 FCC600JACXX:2:1101:1489:2045/1 NGGCAAAAGGAAGCACATATTCGCATATAGAACCAGGATTTATAAGGTACAACAANTAGACTTATCCTCCACTCTCATGTTCATGAATC + #1=ABDD?FH?HFG>DGHBHIFFHGCGGCHGCGHGII)?FG@DBAEH9??FGAB#-5@(@=EEHECAAH@EE;BCEEFA@ADDCCA;AC
2. 按照GC含量进行筛选
按照GC含量的筛选可以按照之前的fasta文件的筛选形式来,而且这个更加简单
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 zcat 123.fastq.gz | perl -n -e ' BEGIN{ # 首先可以定义一个求序列GC含量的子程序 # 这个子程序在之前的fasta文件操作中就已经写过一次了 sub statistic_GC_base{ # 统计序列的GC含量和数量 my $sequence = shift; my $len = length($sequence); my $num = ($sequence =~ tr/GCgc/GCgc/); my $GC = sprintf("%.2f",$num * 100 / $len); # 返回的是百分数(不带百分号) return $GC; } # 先定义一个GC含量的限制 $GC_upper_limit = 50; $GC_lower_limit = 40; } (my $title = $_) =~ s/\r?\n//; (my $sequence = <>) =~ s/\r?\n//; (my $comment = <>) =~ s/\r?\n//; (my $quality = <>) =~ s/\r?\n//; # 比如我这里限制GC含量为50%,小于这个值才能通过 my $GC_percent = statistic_GC_base($sequence); if($GC_percent <= $GC_upper_limit and $GC_percent >= $GC_lower_limit){ print "$title\n$sequence\n$comment\n$quality\n"; } '
说实话可能对于fastq文件进行GC含量的筛选可能意义不大,因为本来序列准确度不一定。有些看起来含量在合适的范围内但是很可能因为测序错误导致的,这里只是说提供了一种思路。
3. 按照质量值筛选
截断
把read的两端的序列按照一定原则进行截断得到质量高的序列
截断两端一定的长度
我觉得这种截取的方式太过于笼统,容易丢失一些信息,然后也可能容纳一些错误信息。
1 2 3 4 5 6 7 8 9 10 11 12 13 export left_len=10export right_len=20zcat 123.fastq.gz | perl -n -e ' (my $title = $_) =~ s/\r?\n//; (my $sequence = <>) =~ s/\r?\n//; (my $comment = <>) =~ s/\r?\n//; (my $quality = <>) =~ s/\r?\n//; substr($sequence,0,$ENV{left_len},""); substr($quality,0,$ENV{left_len},""); substr($sequence,0-$ENV{right_len},$ENV{right_len},""); substr($quality,0-$ENV{right_len},$ENV{right_len},""); print "$title\n$sequence\n$comment\n$quality\n"; '
再加上上面的长度分布统计的代码之后
输出为:
1 2 3 4 length count 59 1 60 18 61 1
根据质量值进行截断
如果从右往左边数,碱基错误率低于20,那么截断它,即使在错误与错误之间有单个质量值高的碱基:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 test_line=80 zcat 123.fastq.gz | head -n $test_line | perl -n -e ' BEGIN{ # 新建一个子程序来判断质量值 sub whether_bloew_20 { my $word =shift; # 这里我默认是Phred+33的模式 if(ord($word)-33<20){ return undef; }else{ return 1; } } # 新建检查两端需要截断长度的子程序 sub side_cut_off { my $sequence = shift; my $direction = shift; # 查看的方向 my $increase_num; if($direction eq "left"){ $increase_num = 1; }else{ $increase_num = -1; } my $num = int(- 0.5 + $increase_num); # 记录目前查看碱基的位置 my $error_base; # 统计不达标碱基的数量 my $right_base; # 统计达标碱基的数量 my $word; while($word = substr($sequence,$num,1)){ # 如果达标的碱基连续达到5个,那么就结束检测 if($right_base >= 5){ return $num; } $num += $increase_num; if(whether_bloew_20($word)){ $right_base++; }else{ $right_base = 0; $error_base++; } } # 说明这条read没救了 return undef; } } (my $title = $_) =~ s/\r?\n//; (my $sequence = <>) =~ s/\r?\n//; (my $comment = <>) =~ s/\r?\n//; (my $quality = <>) =~ s/\r?\n//; # 检查左边 my $left_trim = side_cut_off($sequence,"left"); # 然后检查右边 my $right_trim = abs(side_cut_off($sequence,"right") + 1); # 如果序列没救了就直接跳过 unless($left_trim){ next; } # 去掉低质量 substr($sequence,-$right_trim,$right_trim,""); substr($quality,-$right_trim,$right_trim,""); substr($sequence,0,$left_trim,""); substr($quality,0,$left_trim,""); print "$title\n$sequence\n$comment\n$quality\n"; '
性能怎么样现在还不知道,只是说这里提供了一种思路。
直接丢弃
如果read的质量值或者长度不符合条件那么直接将其舍弃。
参考Expected errors predicted by Phred (Q) scores
也就说这里我们用E值来对read进行筛选:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 zcat 123.fastq.gz | perl -n -e ' BEGIN{ # 将Q值转换为P值 sub Q_to_P { my $q_value = shift; my $p_value = 1/(10**($q_value/10)); return $p_value; } sub convert_quality_to_score{ my $ascll = shift; # 这里我默认使用Phred+33 return ord($ascll) - 33; } } (my $title = $_) =~ s/\r?\n//; (my $sequence = <>) =~ s/\r?\n//; (my $comment = <>) =~ s/\r?\n//; (my $quality = <>) =~ s/\r?\n//; my $total = 0; my $site = 0; my $word; while($word = substr($quality,$site,1)){ $site++; $total += Q_to_P(convert_quality_to_score($word)); } $total = int($total); # 如果有10以上的碱基可能错误,那么就丢弃这个read if($total <= 10){ print "$title\n$sequence\n$comment\n$quality\n"; }else{ next; } '
4. 按照相关性筛选
含有某段特定的序列
这里可以使用grep或者perl的正则表达式:
1 zcat 123.fastq.gz | grep "ATGG"
这个是简单的写法,也可以利用正则表达式:
1 2 3 4 5 zcat 123.fastq.gz | awk 'NR%4==2{print}' | perl -n -e ' if(m/ATG\w+T(AA|AG|GA)/){ print; } '
也可以着色:
1 2 3 4 5 zcat 123.fastq.gz | awk 'NR%4==2{print}' | perl -n -e ' if(m/ATG\w+T(AA|AG|GA)/){ print; } ' | grep 'ATG' --color=always
与某些序列能比对上的
这里实际上不能只使用perl语言,还需要搭配其他工具,这里先不写,占个位。以后补起来。
其实除了筛选这些序列以及质量值之外,还有第一行有很多信息也可供筛选,比如将哪些tail的序列筛选出来,去除哪个tail中的对应位置的序列等等。
检查
1. 检查fastq文件
1 2 3 4 5 6 7 8 9 10 11 pigz -d -c 123.fastq.gz | perl -n -e ' chomp(my $title = $_); chomp(my $seq = <>); <>; chomp(my $quality = <>); if(length($seq) != length($quality)){ print $. - 3 . "lines <$title> is error.\n"; print "You can use\n\n cat *.fastq | head -n " . $. - 4 . "\n\n get the available reads!"; exit; } '
2. 修复
后续添加进来
组合起来!
上面的每一步单独的步骤前后用管道连接起来就可以连续完成多个工作啦!
例如
1 读取fastq文件 | head | 按照长度筛选 | 按照质量值筛选 | 将序列转换为fasta序列
1 读取fastq文件 | head -n 80000 | 将序列转换为fasta序列 | 统计每一条序列的长度
1 读取fastq文件 | head -n 80000 | 按照质量值筛选 | 将序列转换为fasta序列 | 统计GC含量分布
除此之外还有别的组合,按照自己的需要来,上面的步骤中部分用到了之前的fasta文件的相关操作。
有意思的
参考自青蛙快飞
1 2 3 4 5 6 7 8 zcat 123.fastq.gz | perl -n -e ' (my $title = $_) =~ s/\r?\n//; (my $sequence = <>) =~ s/\r?\n//; (my $comment = <>) =~ s/\r?\n//; (my $quality = <>) =~ s/\r?\n//; $quality =~ tr{!"#$%&' ()*+,-./0123456789:;<=>?@ABCDEFGHIJKL}{▁▁▁▁▁▁▁▁▂▂▂▂▂▃▃▃▃▃▄▄▄▄▄▅▅▅▅▅▆▆▆▆▆▇▇▇▇▇██████}; print "$title \n$sequence \n$comment \n$quality \n" ; '
测试数据
因为数据量太大,对于本次的实战演练耗时较长,所以我取了80行的fastq的信息来作为测试用。
新建文件
在windows下新建方法,右键 - 新建 - 文本文档 - 改名为123.fastq
在linux、mac等命令行中新建的方法,打开shell - 输入echo -n 123.fastq
加入数据,将下面的测序数据拷贝到
文件中
在windows下,用记事本打开,拷贝下面的数据然后保存。
在命令行中,那就先vim 123.fastq,打开文件,然后按a进入编辑模式,将下面的数据拷贝到里面。然后按下Esc,输入:wq,按下Enter保存。
压缩为
打开git for windows或者shell,输入gzip 123.fastq。
将下面的数据拷贝到
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 @SRR2177462.1 FCC600JACXX:2:1101:1489:2045/1 NGGCAAAAGGAAGCACATATTCGCATATAGAACCAGGATTTATAAGGTACAACAANTAGACTTATCCTCCACTCTCATGTTCATGAATC + #1=ABDD?FH?HFG>DGHBHIFFHGCGGCHGCGHGII)?FG@DBAEH9??FGAB#-5@(@=EEHECAAH@EE;BCEEFA@ADDCCA;AC @SRR2177462.2 FCC600JACXX:2:1101:1661:2085/1 NAATGAAATTAAAGATAGCTGATCTATATTTCTCAAGTGACTAAGTATTAATATTATGCGTACTCTGTATTTCTCTAGTTGGTGGTTTAG + #4=DDFFFHHFHFIGIHIIIJEGHIGIAHIJFIEHHHIIIIIEIHIIIJCFIHJIHIIAHIFFGIIJHFHIIJIJF@FEHGIDHHA?B@C @SRR2177462.3 FCC600JACXX:2:1101:1683:2113/1 GCGACCTCGCGGGCGAAGCCCATCTCGTGGGTGACGCAGATCATCGTCATGCCCTCGGCGGCGAGGCCTTTCCTGACGGCGGGGACCTCC + @@@BDDDDFHDDA:DDBHH@F6=DCEHHHFE6>;;?86?BC:ACCC@98?######################################## @SRR2177462.4 FCC600JACXX:2:1101:1730:2127/1 TTTGTCCACTTCCTCCACTTCGTCAACTTCGTCCACTTCGTCCACTTCCTCAACTTCGTCAACTTCGTCCACTTCGTCAACTTCGTCCAC + BBCFFFFFHHHHHHJJJJIJIIJJEIHHHGIJHIJJIIGGIGGIJGHDFHC4BBCGC@G;(;CEGEEH??@DCFD?CC=>CDDDDDD??D @SRR2177462.5 FCC600JACXX:2:1101:1663:2160/1 ACAAAAAGCACAGAGAAGCATTGAGAATGGGGGATTTTGGAGAGCTCCAGGGGGACCTTCATCTTCACTGGATCATCATCCATTGCCAAG + @BCFFFFFFHHFHGHIJJHGHIIFHGCHIJJJJBHGIJHGHGGGIHFHFHCGIHBCDECCCDDDDCDCCDBCDCB>CC@CCDDEDDCCDD @SRR2177462.6 FCC600JACXX:2:1101:1555:2174/1 CACAATTGGTGAGTTCTAACGTTAGCATTATTGGTTTGAAAGTATATATGGAACACTTATACCTAGAACTATCGTTTTTGTCATTATTAA + @@@FFFFDHAFFFFBEGGIIIFHDGHEDHIII>FFHHIGGGGBFHGIIIGHECBGDGHGGCAG@G==FCF=FAC=@ACE/6?BEFDCCB@ @SRR2177462.7 FCC600JACXX:2:1101:1561:2201/1 TCTCCTTTAGCATCAAACCCTCCGTTGGAATAACCCAACAACCATACATACATTCATAGAGAAAACCCAACACAAATATTACATAAACCT + @@@BDDDA<?ADF@DHGGCD9:A@AGE??EC?9:???BFHI)8BFEFIIG8=CHG)=FA@4@GEGII;AABDEB;;@CDC>CCDCCCC5? @SRR2177462.8 FCC600JACXX:2:1101:1676:2217/1 GCTAGTTCCTAACAGAACTCGAGGTAACAGTCCAAGGGATTGAAAAAACACAAGCTGATGTTCGTCGAAGTAATACAACAAGAAGGAGAC + @CCFFFFFFHHDDBHIIGI?E@GHCGHGGGHIB>DECEBBFCABEDGGEECHIGGGACHG;@AEE?DBBB5>CADA>(;99<CCCB9?A@ @SRR2177462.9 FCC600JACXX:2:1101:1700:2228/1 CAAAACACTTCAATAGCATGTTCTAAAAGGACTGCTTTAAAAATCTTCTACAAATCTCACATGTTTCAAACTAGATTTTGAAGCAATAAA + CCCFFFFFHHHFFIIJIIGIIIDHJJJIHJEHIIHIIIFHHIEHIIIIJGGHEHFGIJIIJIJHIJJBFHJIJJIHIJJJEEEEFFFFFF @SRR2177462.10 FCC600JACXX:2:1101:1962:2055/1 NTTATGTGATGTGAATGATTATGTATAATTGTATGAAACTTAGTAAATTAATATATGATTAATCTTGTTTAAACTAGCTTATCCTATTGT + #4=BDFDFFHHHHGIIJHJJGJJJIIIHHJJIIIIHIJJIJGHHGIJIJHIJJJIJJJJJIIIJJJJIJJHHGIIJIJJJIIGJJHJJJJ @SRR2177462.11 FCC600JACXX:2:1101:1774:2118/1 CACCAAGACGCTACCTAAGGATAGATTTTTTGAGCTTAGAAGAGAGTTAGGAGTATTAGACATGTCTTAGGATCAACGATTATGAAAATTT + CCCFFFFFHHHHHIGIGGGHEHEHHIJJJJJJ?GGHIGIIJGGIHIGIIJDFH7@EHIIGHHGHAHHFDEFFCEE>CC=ABDDDCDDDDCD @SRR2177462.12 FCC600JACXX:2:1101:1931:2123/1 CAGCTTTTTGTGGTTTGCAAGGAAACTGAGCAGAAATATGAACCACATGTGACAACAAAAAAATTAGGAAGCTGTCTGTGAATTATTATT + @@@DFFFFHHFFHGIJIGGEHIBGHIJIDHHJJFIEIIJJJGHIIGIIFGHGIIJJIIJIJIHFDF=D>CEDEECCACCC>AFDDEACDD @SRR2177462.13 FCC600JACXX:2:1101:1759:2210/1 AATAAATGTGAAAATGGCAATAATAACAAACCTGCTGCAATTATACTTGTTATCTTTATCCAAGATTTCAGGTGCAGTAAATTGTCCCAG + @CCFFFFFHHGGHIJJJJJJJIEIJJIIJJJJJJJJJJIJJJJJJJJJJIIIIIJJJIJJGJIJIJJIJGJJHIJHIIJIGHHHHHGEEF @SRR2177462.14 FCC600JACXX:2:1101:1922:2245/1 TATCATTAAACAAACATGCACATTGCTTAAGAAACATAAAACTTCCACTGAAACAATTTATAAGATTGCACCAGATTTATTCCTTTTTGT + CCCFFFFFHHHGHIJJJJJJJJJJJJJJIIJJJJJJJJJJJJIIJHIJIJHJIJJJJJJGIJJJJJJJJIJIJJADHIHHHHHHHFFFFE @SRR2177462.15 FCC600JACXX:2:1101:2088:2085/1 NATGACGTATATTAAGTGTTGAAGATGAAGACATATGTGGTTGGTTTGTTTATGTGTATTTCTCTCTCTGTGCTTCGTTTATGCTTCATT + #1=DDFFDHHHHHIIJGHJJJIIHIGIHGHHHIJIIIHIIFHIJ?GHIGHIBHGICBAHEIHIIJJIJJIJJIIJJHHFFFFFFFEDDEE @SRR2177462.16 FCC600JACXX:2:1101:2166:2127/1 TGGAGGAAGTGGAGGAAGTGGAGGAAGTTGACGAAGTGGACGAAGTGGAGGAAGTGGACGAAGTGGAAGAAGTGGACGAAGTGGAGGAAG + @CCFFFDFHDFHHHJGGIEEHBHIIJJIAGGHIEHI8DHDHGGFH;CGAEE;DGECHC)99>A########################### @SRR2177462.17 FCC600JACXX:2:1101:2243:2189/1 CTTGTCAGAACCCATTCAAAACCACTAAAAACCCCTGCATAGTGGAAGCCAAATGTTAAGGAAGAGAGCTGGAAATCAGAGAGTGGAAAA + CCCFFFFFHHHHHIJJGIJJJIJJIJJJJJJJJIJJJJJJJJIJJDHHHIGIIIJIII@GIIJIJJIHFHHFFFF@EDEEEEDACD@@AC @SRR2177462.18 FCC600JACXX:2:1101:2145:2249/1 AATATTTGATCAATAGATTTTATGATAAATATAAACATACAGTAGAATTTTAAAAAACAAGGAATTTAGCTACCAAAAACAAGCTATGAG + <<@AABDD>HHFBFH@EHIHIADHIEGEHIHDAEHH<FFGBFEGDFHIIHGA>BGB@FFFEDCGGHIGIIEG@DECCHHGFDC?C@EEEE @SRR2177462.19 FCC600JACXX:2:1101:2366:2232/1 TACTGAACTTCTTCAAGAATTTGATCAACCCAGGAACCTATTGCAATGTCTCAATCATAGGAACTTTAATCTCCAATTTCTTGAAGATTT + CCCFFFFFHHHHHIJJJJJJJJJJJJJJJJJJJJIJJJJJJJJJJGHJIJJJJHIJDHIJJGIJJJJIHIGIIIIGIGHGHHHEEFDFFF @SRR2177462.20 FCC600JACXX:2:1101:2455:2241/1 CACGGATCTGCAGAATAGCACCAAAGCTGGAGCTATCACTTTACTTCGTCAACTTCGTCCACTTCCTCCATTTCTTCCACTTCCTCCACT + @@CFFDFFHHGGGIJJJJJJJJJJJJIJJJHIJIJJJJJIJJGJJJGGHIIJIIEEHGHEIIIJIHHHEEHFDEFBDBCE>>ACCDCCDC
后记
其实现在有很多软件可以处理fastq文件,比如FASTX-Toolkit、seqkit、FaQCs、fastqc、fastp、Reseqtools等等,也不需要perl写几行脚本来处理。那这个还有什么意思呢?
你要是读了之后,你会发现在文中我提到了有关代码重用的问题,实际上在处理这类文件的时候有些东西是相通的,你可以在这个文本里面中会看到之前我在操作fasta文件的时候用到的代码。而把这些代码拿到这里来用照样可以达到目的。这个过程就有点像搭积木,积木的基本形状也就那几种,什么正方体、长方体、直角等腰三角形、圆柱等灯,可是不同的搭配、不同布局搭起来的东西就不一样了。当我们把这种单一的,完成某种独立功能的代码组合起来之后就会由一块一块的积木搭建起整个城堡,迎接你的公主!
除了代码之外,在文中也说明了有关质量值编码的问题,话说现在都是采用的Phred+33的编码方式了,那还提那些陈年旧事有什么意思了?有的时候我也在想在课本中提到的那些年代久远的故事究竟有什么作用呢?比如生化书上面的那些经典的比如三羧酸循环,那些都是几十年前就已经搞很清楚的事情为什么还要提呢? 我觉得仁者见仁智者见智吧!可能我有点怀旧吧!
参考
链接
引用



 微信打赏
微信打赏